
- Next-Gen Sequencing and disease

- long non-coding RNAs and the role in cell differentiation
- Predicting novel cis-regulatory modules with prior knowledge of related CRMs
Most of the current CRM discovery approaches rely on the knowledge of related motifs. They normally search the genomic regions for clusters of such motifs. Thus, their accuracy is bounded by how well, if at all, the motifs are characterized. Other methods simultaneously search for motifs and CRMs, but the “unsolved” nature of the motif discovery problem cast doubts upon the scalability of such methods.
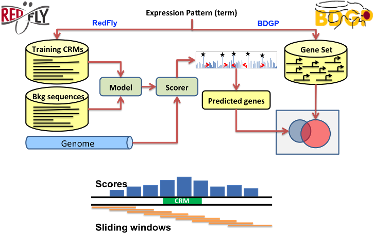
Supervise CRM prediction pipeline
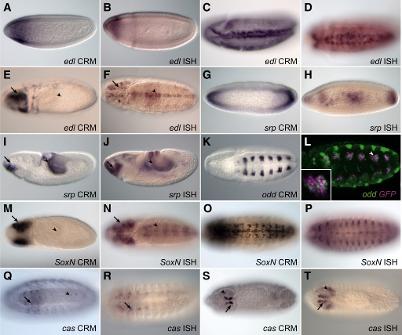
Experimentally validated fruit fly CRMs
Thanks to the extensive work of biologists over the last few decades, who have tested many sequence fragments for regulatory activity in a reporter gene assay, we have now an invaluable collection of known enhancers in variety of species and tissues.
Our goal here is to use a small set of known CRMs participating in a transcriptional network as “training data” to guide the search for other CRMs with similar functionality in the network. We call this task “supervised CRM prediction”. To this end, we constantly develop novel statistical/probabilistic models to capture the similarity between any given sequence and the training data set. We employ our models to locate the high scoring regions of the genome that are potential candidate CRMs of the same network. Experimentally validated candidates in fly and mouse confirms the power of these techniques.
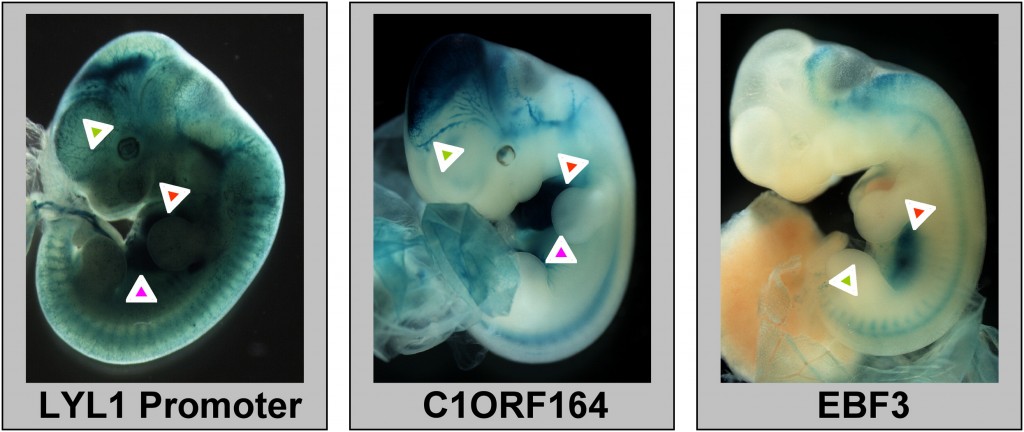
Experimentally validated human CRMs in mouse
- Modeling gene/CRM expression pattern
Finding when, where and how intensely genes are expressed is a challenging problem. Thermodynamic models address some aspects of the problem in fruit fly anterior-posterior (A/P) segmentation network by modeling the expression pattern in nearly 50 of its known CRMs (as training data) during the embryonic stages. These models can be invoked to predict the expression profile of any sequence, given the necessary context. However, such models are too slow to be used in a genome-wide search for CRMs that recapitulate their nearby gene’s expression.
Here, we develop a model that maps the binding sites and the concentration information of relevant transcription factors to the expression pattern driven by several known CRMs involved in anterior-posterior (A/P) patterning. We deliberately sacrifice a negligible amount of accuracy for simplicity and efficiency by replacing the thermodynamic model with a logistic regression that has fewer parameters.
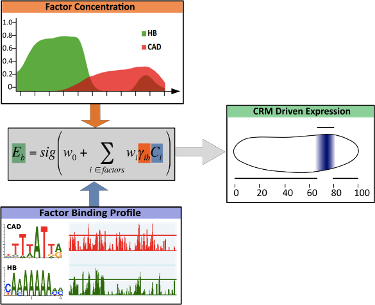
Modeling the expression pattern
Cis regulatory module discovery
Predicted regulatory network
Our model enables us to not only search genome-wide for other CRMs of the network, but also provides a simple mechanism to statistically infer the effect of each TF on each CRM. The video on the left shows how the model is used to scan the region around a gene (e.g. hkb) for segments that could drive expression similar to that of the gene. The middle panel plots the expected and predicted expression pattern for each window, and the bottom panel plots the similarity between expected and predicted expression patterns for that window.
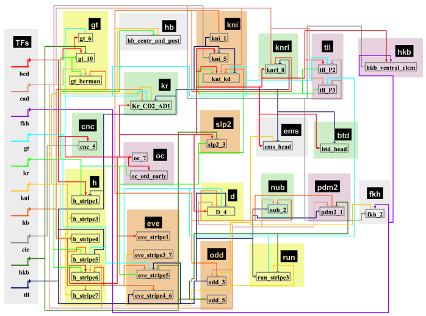
- Transcription factor interactome (iTFs)
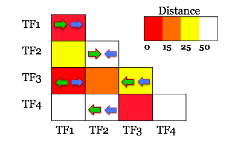
TF-TF interaction signature
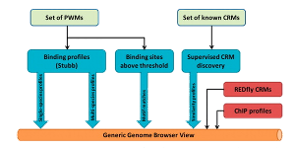
Genome Surveyor scheme
- Annotating genome (Genome Surveyor)